 利用可持续方法获得绿色能源的电催化分解水制氢
利用可持续方法获得绿色能源的电催化分解水制氢
【研究背景】
电催化分解水制氢是利用可持续方法获得绿色能源的有效策略。通常将Pt、Ir或Ru基金属分别用作析氢反应 (HER) 和析氧反应 (OER) 的电催化剂。然而,对于大规模电解水,上述催化剂达到200-500 mA cm-2的高电流密度仍然需要非常高的电压(1.8-2.4 V)。此外,贵金属元素的稀缺性、高成本和相对较低的电化学稳定性限制了其商业前景。因此,开发高成本效益、基于丰富的原材料、贵金属用量超低的催化剂非常关键。
具有不饱和位点的铁钴层状双氢氧化物 (FeCo-LDH) 催化剂能在碱性条件下同时实现HER和OER。为了满足低电压(<1.8 V)下大电流密度( 500 mA cm-2)的工业要求,必须进一步改进电解水催化剂。为了提高LDH的催化活性,一种经典的方法是通过打破催化剂表面结构的对称性,调整界面处的相互作用、配位环境和电子性质,促进电荷转移。金属-载体相互作用可以进一步提高本征催化活性。
500 mA cm-2)的工业要求,必须进一步改进电解水催化剂。为了提高LDH的催化活性,一种经典的方法是通过打破催化剂表面结构的对称性,调整界面处的相互作用、配位环境和电子性质,促进电荷转移。金属-载体相互作用可以进一步提高本征催化活性。
一种有效的策略是通过交替的Fe和Co原子排列,控制LDH生长;同时通过掺杂活性原子,构建多原子异质界面。活性金属原子进入LDH的骨架结构,破坏其有序原子排列的对称性,导致不饱和键断裂,并伴有局部电荷过剩,可能对催化活性产生积极影响。更重要的是,掺杂后得到的LDH以另一个原子作为新的活性位点,可以有效增加活性位点的数量,并诱导其杂原子界面处的电荷转移,从而调整反应吸附能,并调控分解水的性能。部分取代效应可以抑制工业反应条件下多杂原子界面的不稳定性。然而,如何打破多杂原子界面的对称结构,并构建基于LDH的稳定多原子异质界面,仍具有挑战性。
【成果简介】
武汉理工大学的木士春教授和南京晓庄学院的刘苏莉教授(共同通讯作者)在铁钴层状双氢氧化物 (RuxSACs@FeCo-LDH)上构建的Ru单原子催化剂在10和1000 mA cm-2的电流密度下表现出194和246 mV的极低析氧反应 (OER) 过电位,在1000 mA cm-2连续反应1000 h后,表现出极高的稳定性,其质量活度分别是Ru和FeCo-LDH的2倍和6倍。
【研究亮点】
1. 在1.52 V电压下,在电解水反应中可实现高达1000 mA cm-2的电流密度;
2. FeCo-LDH表面的结构对称性被破坏,在电催化条件下原位形成了类似于Ru-O-TM(Fe、Co 和 Ni)的纳米级化合物,促进O-O耦合。
【图文导读】
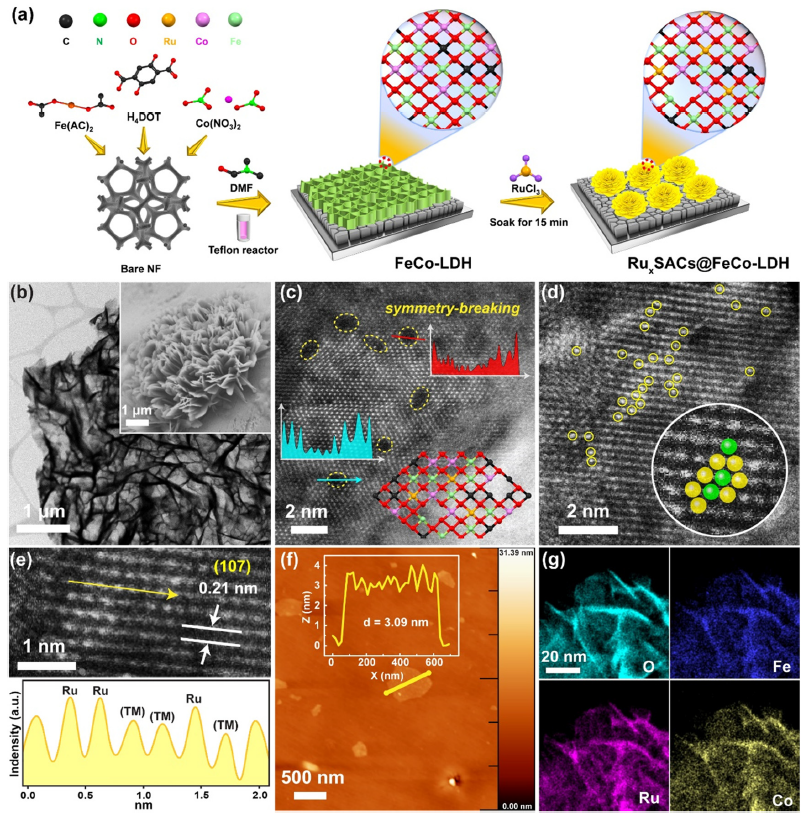
图1 (a) 水热合成电催化剂,(b) Ru1SACs@FeCo-LDH的TEM、FE-SEM和HAADF-STEM,(c)黄色虚线圈出的缺陷,(d) 明亮的黄色圆圈标记的点代表单原子Ru,(e)图像中白线之间的强度分布 (TM = Fe, Co),(f) AFM,(g) Ru1SACs@FeCo-LDH的元素映射图。
作者使用混合溶剂策略,用水热法合成RuxSACs@FeCo-LDH催化剂。如图1a所示,使用泡沫镍(NF)作为基底,先生长FeCo-LDH阵列,然后原位生长Ru单原子,从而打破了FeCo-LDH阵列原有的对称性,有利于催化反应。TEM和FE-SEM图像显示,Ru1SACs@FeCo-LDH仍然保持了由互连的超薄纳米片组成的均匀3D多孔结构(图 1b)。
如图1c所示,HAADF-STEM图像显示了催化剂表面对称性被破坏后的原子结构,这意味着在LDH表面形成了包含多种原子的界面,从而增加了活性表面积和活性位点数目。观察到的亮点(图 1d)对应于单个Ru原子,线性扫描分析进一步证明了Ru原子的孤立状态(图1e),但由于原子序数接近,无法区分Fe或Co原子。图1f中,Ru1SACs@FeCo-LDH纳米片厚度仅为 3.09 nm。能量色散 X 射线 (EDX) 光谱进一步证明了 Fe、Co 和 Ru 物种在FeCo-LDH纳米片上均匀分布(图 1g)。
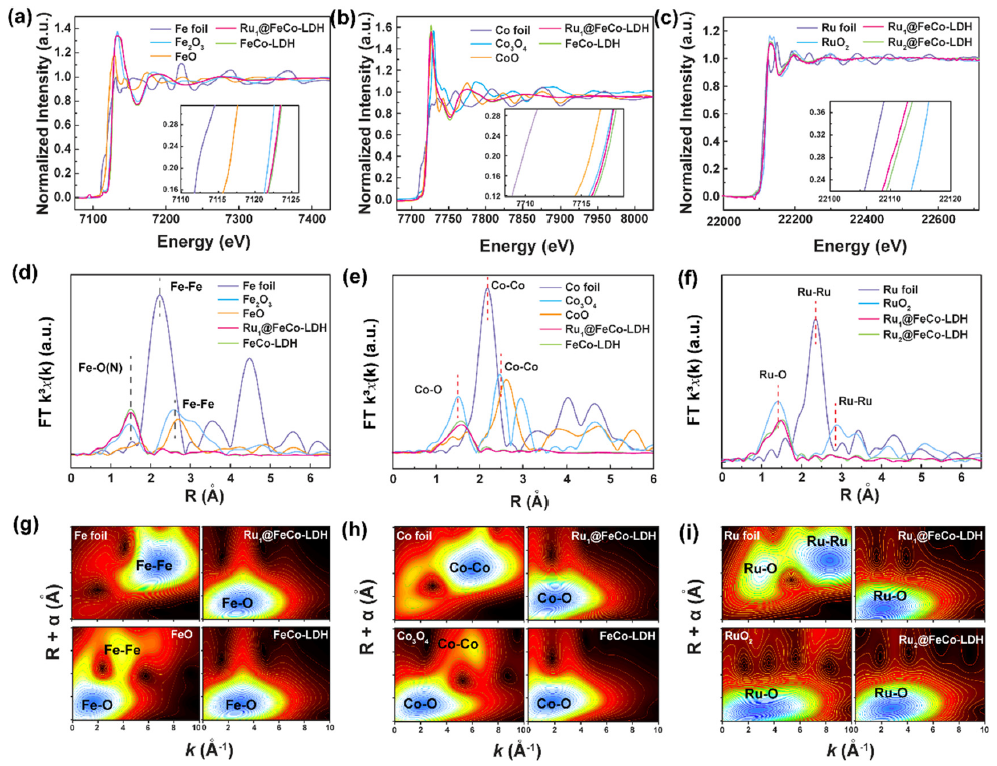
图2 Ru SACs@FeCo-LDH 的(a) Fe, (b) Co和 (c) Ru的K-边XANES光谱。(d) Fe 箔、Fe2O3、FeO、Ru1SACs@FeCo-LDH和FeCo-LDH的R空间傅里叶变换 (FT)。(e) Co箔、Co3O4、CoO、Ru1SACs@FeCo-LDH和FeCo-LDH的R空间FT。(f) Ru箔、RuO2和RuxSACs@FeCo-LDH的R空间FT。(g–i) Co、F和Ru 的 K 边 EXAFS 光谱的加权函数。
Ru1SACs@FeCo-LDH和FeCo-LDH样品的Fe XANES光谱在~7133.2 eV处显示出明显的前边缘峰(图2a),表明原子级分散的Fe物种呈+3价。与FeCo-LDH 相比,Ru1SACs@FeCo-LDH 显示出向较低光子能量的轻微转变,证实了Ru-O-Fe结构的形成。与FeCo-LDH相比,Ru1SACs@FeCo-LDH的Co K 边吸收边显示出相同的轻微负移,证明了价态介于+2和+3价之间的Co物种(图 2b)。图 2c表明,Ru的氧化态在0和+4价之间,并证明Ru原子带正电荷。如图2d所示,Fe元素的EXAFS光谱的傅里叶变换显示R = 1.5 Å 处的峰,对应于Fe-O。
对于Ru1SACs@FeCo-LDH,由于Fe和O在LDH结构中的结合,只能检测到Fe-O散射的信号,因此排除了Fe、Fe2O3的存在。Ru1SACs@FeCo-LDH的Co K边位于1.6 Å(图 2e)。图 2f中的FTR空间光谱表明,RuxSACs@FeCo-LDH 没有Ru-Ru键的散射峰,这反映了Ru1SACs@FeCo-LDH和Ru2SACs@FeCo-LDH中存在单原子分散的Ru。如图2g-i所示,RuxSACs@FeCo-LDH仅在 2.4 Å 处出现 Fe (Co, Ru)-O 键的特征峰,可以归属于原子 Fe ( Co, Ru)-O 物种。Fe-Fe、Co-Co和Ru-Ru配位的缺失消除了Ru簇存在的可能性。
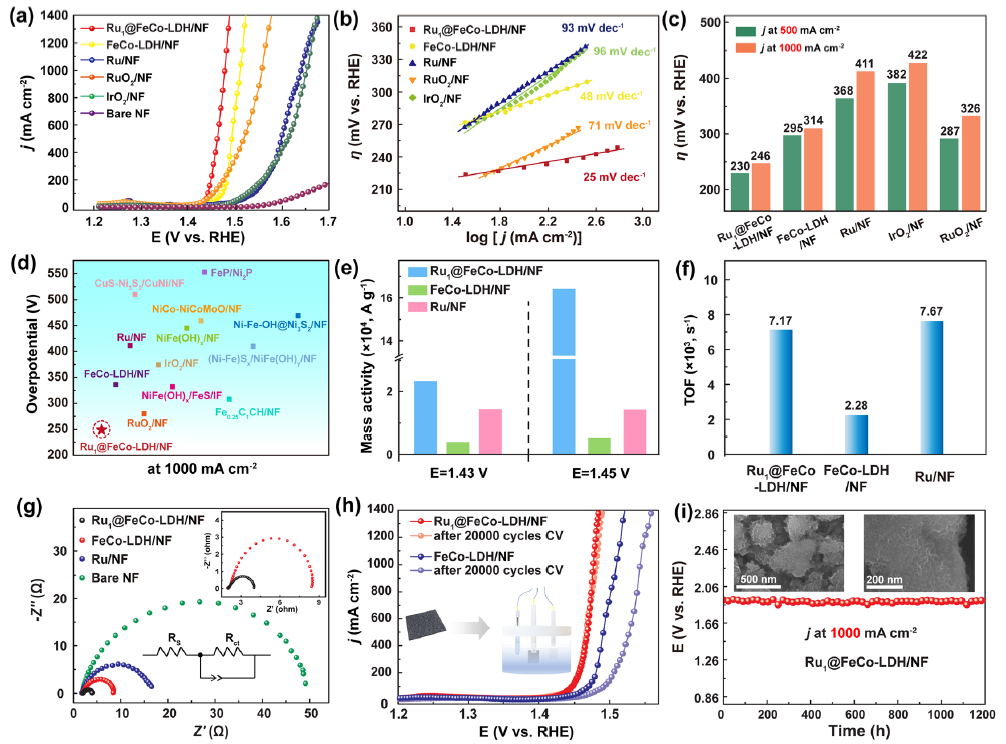
图3 (a) 1.0 M KOH中的极化曲线;(b)塔菲尔曲线;(c)在500 mA cm-2和 1 A cm-2下的过电位比较;(d) 在 1.0 M KOH 溶液中,与最近报道的1 A cm-2下的OER 催化剂过电位对比;(e) 不同过电位下的质量活性比较;(f) 不同电位下的TOF;(g) 奈奎斯特图;(h) Ru1SACs@FeCo-LDH和FeCo-LDH在20000圈CV循环前后的极化曲线;(i) Ru1SACs@FeCo-LDH在1 A cm-2电流密度下连续反应1200小时所需的电位随时间演化的计时电位曲线。
如图3a 所示,空白NF达到500 mA cm-2的电流密度需要439 mV的过电势。掺入单原子Ru后,Ru1SACs@FeCo-LDH表现出最高的OER活性。Ru1SACs@FeCo-LDH呈现出25 mV dec-1的最小Tafel 斜率(图3b)。图 3 c证明了 Ru1SACs@FeCo-LDH具有优异的OER催化活性。这比大多数先前报道的催化剂更好(图3d)。
对于Ru1SACs@FeCo-LDH,其在 200 mV 过电位下的质量活性(MA)分别是FeCo-LDH和Ru的6倍和2倍(图3e)。当过电位为200 mV时,转换频率 (TOF) 为7.17 × 103 s-1 (图 3f )。EIS证实了 Ru1SACs@FeCo-LDH 的快速反应动力学(图 3g)。如图3h和i所示, Ru1SACs@FeCo-LDH在1 A cm-2的电流密度下连续反应1200 h后,活性仅下降5%,表明其具有良好的稳定性。经历1200 h的OER后,HRTEM没有观察到明显的形貌变化。
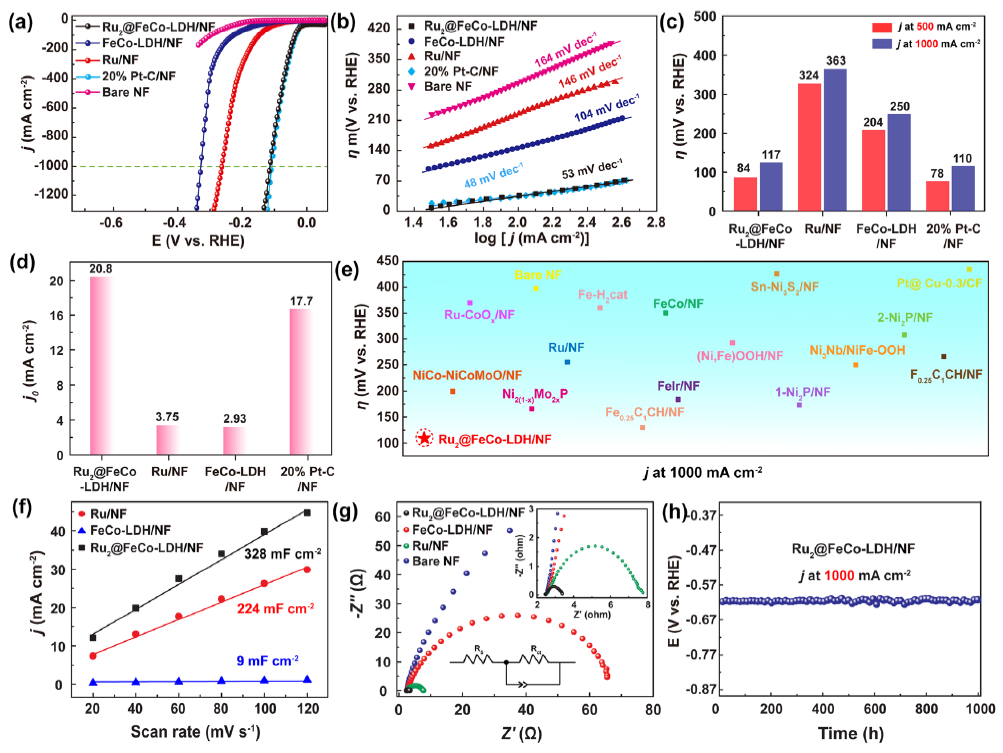
图4 (a) 1 M KOH中的极化曲线;(b)塔菲尔曲线;(c) 在 500 mA cm -2 和 1 A cm-2下电催化HER的过电位;(d) 交换电流密度 (j0);(e) 在 1.0 M KOH 溶液中达到1 A cm -2的电流密度时,与最近报道的HER 催化剂的过电位对比;(f) Cdl;(g) 奈奎斯特图;(h) Ru2SACs@FeCo-LDH在1 A cm-2下连续反应1000小时的计时电位曲线。
如图4a所示,Ru2SACs@FeCo-LDH 在碱性介质中表现出优异的HER活性, Tafel斜率低至53 mV dec-1(图 4b),接近商业 Pt/C 催化剂。如图4c所示,在500 mA cm-2和 1 A cm-2的电流密度下,过电势分别为 84 和 117 mV。与对照组催化剂相比,Ru2SACs@FeCo-LDH表现出最大的交换电流密度 (j0 ) (图 4d)。
Ru2SACs@FeCo-LDH的活性高于大多数报道的电催化剂(图 4e)。图 4f 表明,Ru2SACs@FeCo-LDH 显示出最大的Cdl和最小的Rct (图 4g),这表明快速的HER动力学来自增大的电化学表面积和显著提升的电荷转移能力。Ru2SACs@FeCo-LDH表现出优异的电化学稳定性(图 4h)。
图5 (a) 全分解水装置;(b) 双功能RuxSACs@FeCo-LDH电催化剂的极化曲线;(c) Ru1SACs@FeCo-LDH的产气量;(d) RuxSACs@FeCo-LDH在1 A cm -2下连续反应1000 h的计时电位曲线;(e) RuxSACs@FeCo-LDH与报道的双功能电催化剂在500 mA cm-2和1 A cm-2下所需的电压对比。
作者组装了如图5a所示的碱性电解槽。与基于其他电催化剂的电解槽相比,在1.0 M KOH溶液中,该碱性电解槽表现出最佳的全分解水活性(图 5b)。从图5c可见,收集的H2和O2之比约为 2:1,法拉第效率接近100%,具有优异的耐久性(图 5d),可用于工业规模的全分解水催化剂(图 5e)。

图6 (a,b) RuSACs@FeCo-LDH和FeCo-LDH的Fe、Co和Ru活性位点的投影态密度。(c) FeCo-LDH和(d) Ru SACs@FeCo-LDH电催化OER过程中的自由能变化。(e)Δ GOOH*、 ΔGOH*、 ΔGO*和d带中心之间的比例关系。(f)作为d带中心函数的过电位火山图。(g) FeCo-LDH和Ru SACs@FeCo-LDH电催化OER的机制。
如图6a和b所示,Ru SACs@FeCo-LDH上,Co活性位点的d带中心比FeCo-LDH更接近费米能级,表明Ru SACs@FeCo-LDH在电化学反应过程中具有更强的电子供受能力。随着d带中心能级的提升,OER中间体的吸附能力增强,理论上可以提高FeCo-LDH的OER活性。基于FeCo-LDH的自由能图(图6c), O2脱附是Co位点催化下的OER速控步骤。
将Ru引入FeCo-LDH后,Ru位点(图6d)增强了OER中间体的吸附能,即通过负移的d带中心优化了中间体的吸附自由能,从而促进了OER活性(图6e)。Ru掺杂降低了OER过电位(图 6f)。与 FeCo-LDH和Ru SACs@FeCo-LDH界面上的Co位点相比,活化能显著降低(图 6g)。
【总结与展望】
为了提升高电流密度下的全解水性能,该工作构建了强耦合单原子(Ru)改性的铁钴层状双氢氧化物(Ru SACs@FeCo-LDH),获得的RuSACs@FeCo-LDH在OER和全解水反应中表现出优异的电催化活性和稳定性。实验分析和DFT计算结果表明,由于FeCo-LDH被亲氧的Ru原子部分取代,打破了杂原子界面的长程有序,即打破了界面原子结构的对称性,进行了原子尺度重构,导致单原子Ru和FeCo-LDH之间形成了强耦合的肖特基界面。
审核编辑:刘清
-
DFT
+关注
关注
2文章
231浏览量
22709 -
傅里叶变换
+关注
关注
6文章
441浏览量
42590
原文标题:木士春&刘苏莉EES:打破单原子催化剂的结构对称性,获得高电流密度、低能垒和高稳定性的全分解水催化剂
文章出处:【微信号:清新电源,微信公众号:清新电源】欢迎添加关注!文章转载请注明出处。
发布评论请先 登录
相关推荐
实时原位监测光电催化过程中反应物浓度与热效应的微光纤传感器技术
快速启停!AEM在新能源制绿氢应用中的适配性。

如何确保为绿色氢能提供高效稳定的 DC 电流
华为超充亮相2024东盟可持续能源周
隆基氢能获得DEKRA德凯认证证书,加速推动能源转型
参加研讨会,下载白皮书 | 揭秘功率转换技术如何重塑绿色氢能

直播预告 | 驱动绿色未来:功率半导体在制氢电能转换的革新解决方案

合理设计具有成本效益的金属掺杂ZrO₂提升OER催化活性

高性能电压巡检方案在电解水制氢行业的应用

励展博览集团宣布收购世界氢能峰会主办方——可持续能源会议(SEC)
氢电动汽车:可持续交通的未来?
村田中国氢能源汽车正式投入运营,持续发力打造绿色低碳供应链

相调控对镍锡合金的电催化氮还原调控机制研究





 工商网监
工商网监
评论